
Behandling
Børn og voksne med cystisk fibrose bruger 1-3 timer om dagen på livsnødvendige behandlinger foruden indlæggelser og månedlige hospitalskontroller.

Herunder kan du læse om lungernes opbygning, og hvordan genfejlen ved cystisk fibrose påvirker lungerne.
Lungerne består af to systemer; det øvre system og det nedre system. Det øvre system består af luftrøret og de store og små bronkier, hvis funktion er at lede luften frem og tilbage. Det nedre system består af alveolerne, hvor selve luftskiftningen til og fra blodet foregår.
Overfladen af cellerne i lungerne er dækket af et lag vand. Ovenpå vandlaget ligger et lag mucus (slim, sekret), som fanger bakterier, der forvilder sig ned i lungerne. I vandlaget står de små cilier (fimrehår) og bølger frem og tilbage i en jævn rytme. Ciliernes bølgebevægelser skubber mucus, der flyder i vandlaget mod de store luftveje, så man automatisk renser luftvejene og kommer til at hoste, når det er nødvendigt. Dette er en del af det naturlige immunforsvar i lungerne.
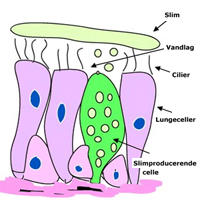
Når man har cystisk fibrose, er vandlaget på overfladenaf cellerne i lungerne meget tyndt og utilstrækkeligt. Laget af sekret (mucus) ligger derved ovenpå cilierne og presser dem ned, og cilierne kan ikke arbejde optimalt.
Fordi vandlaget er utilstrækkeligt, bliver sekretet ikke fortyndet som normalt. Det bliver i stedet sejt og tyggegummiagtigt. Inhalerede bakterier får derved nemmere ved at sætte sig fast på overfladen af slimhinden og er vanskelige at få transporteret væk, da cilierne ikke kan arbejde optimalt pga. det tynde vandlag.

Det seje sekret giver gode vækstbetingelser for alvorlige bakterier. Dette medfører hyppige lungeinfektioner, som man behandler med antibiotika. For at forebygge lungeinfektioner skal børn og voksne med cystisk fibrose flere gange dagligt gennemgå en omfattende hjemmebehandling med slimløsende inhalationer og lungefysioterapi for at bringe det seje slim op fra lungerne. Ved de månedlige kontroller på Cystisk Fibrose Centrene afleveres en spytprøve fra luftvejene, som bliver undersøgt for, om der er bakterier, der skal behandles.
Hvis bakterierne ikke bliver behandlet, kan der dannes arvæv i lungerne, som over tid mindsker lungekapaciteten. På et tidspunkt kan det ske, at det ikke længere er muligt at udrydde bakterierne helt. Så kalder man infektionen for kronisk, og man forsøger i stedet at holde bakterierne nede. Dette sker bl.a. ved antibiotikabehandling, som i nogle tilfælde skal gives intravenøst (iv-kur). En iv-kur varer som regel 14 dage. Første gang man skal have iv-kur foregår det under indlæggelse, men de fleste lærer at tage behandlingen selv, og så er det kun nødvendigt at komme til opstarten af behandlingen.
Læs mere om medicin og behandling her.
Se også denne video, hvor vi illustrerer, hvordan cystisk fibrose påvirker lungerne:
Har du cystisk fibrose eller er du nær pårørende? Vi er her for dig! Som medlem af Cystisk Fibrose Foreningen kan du benytte vores mange medlemstilbud og rådgivning – og du er samtidig med til at støtte vores arbejde for bedre og længere liv for alle med cystisk fibrose.

Børn og voksne med cystisk fibrose bruger 1-3 timer om dagen på livsnødvendige behandlinger foruden indlæggelser og månedlige hospitalskontroller.
Du kan støtte Cystisk Fibrose Foreningen på flere måder:

Brug Cystisk Fibrose Foreningen. Vi har nemlig en lang række tilbud til dig.

Forskning er nøglen til ny og bedre behandling for børn, unge og voksne med cystisk fibrose.


