Et barn har kun cystisk fibrose, hvis det har to mutationer, en fra hver af forældrene. Cystisk fibrose er en sygdom, som ses hos kaukasere (hvide mennesker), men hyppigheden af de forskellige mutationer varierer verden over, alt efter folkeslag. Den hyppigste af alle mutationer kaldes deltaF508.
DeltaF508 på verdensplan
På verdensplan har cirka 85% af patienterne med cystisk fibrose deltaF508-mutationen på mindst ét gen fra forældre. Cirka 47% har mutationen på begge gener. Når man har mutationen på begge gener, kaldes det at være deltaF508 homozygot. Cirka 39% har deltaF508-mutationen på det ene gen og en anden mutation på det andet gen. De er såkaldte deltaF508 heterozygote. Cirka 15 % har andre mutationer.
DeltaF508 ses især i de lande, som tidligere har været europæiske kolonier, eksempelvis Australien, New Zeeland og USA. I USA ses mutationen også blandt sorte som følge af blandede ægteskaber og slaveri. Den ses blandt ‘hispanic’ amerikanere, hvor den er indført fra Spanien. Men den ses ikke blandt ‘pacific’ amerikanere på vestkysten eller blandt ‘eskimo’ amerikanere mod nord.
DeltaF508 i Danmark
I Danmark er forekomsten af deltaF508 meget høj. Der er registreret godt 40 forskellige cystisk fibrose-mutationer i Danmark. Mange af mutationerne optræder kun én gang. Cirka 96 % af alle danske patienter har en eller to deltaF508 mutationer. Det betyder, at de har det, man kalder en klassisk alvorlig og behandlingskrævende cystisk fibrose med generelle komplikationer i både lunger og mavetarmsystemet. Alle disse patienter hører hjemme i mutationsklasserne I, II og III. Over 80 % af patienterne har to deltaF508 mutationer. De hører hjemme i mutationsklasse II. Det er kun cirka 4 % af danske patienter, der ikke har nogen deltaF508 mutationer. Halvdelen af disse har mutationer, som er af ikke-europæisk oprindelse.
DeltaF508 fra land til land
Andelen af patienter (96%) med mutationen deltaF508 i Danmark er usædvanlig høj sammenlignet med andre lande. Herunder ses nogle eksempler:
Norge: 66,7 %
Sverige: 73,3 %
Tyskland: 70,7 %
Frankrig: 60,3 % for hele landet, mens andelen i det vestlige Frankrig er helt oppe på 81,2%
Israel: 32,2 %
Færøerne: Stort set alle patienter med cystisk fibrose på Færøerne har deltaF508.
Variationen bestemmes af landets demografi og ikke mindst geografi.
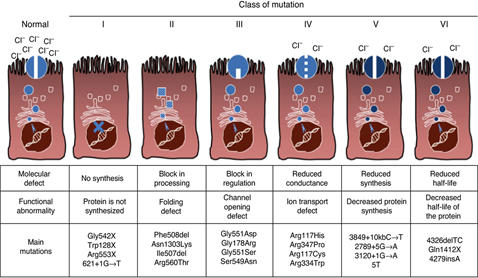
De 6 mutationsklasser ved cystisk fibrose
Cystisk fibrose inddeles i seks mutationsklasser alt efter, hvilken defekt for dannelsen af CFTR-proteinet og dermed CFTR-kanalen, som mutationerne medfører.
Mutationsklasserne er med til at give en forklaring på nogle af variationerne af sygdommens sværhedsgrader, og mutationsklasserne er udgangspunktet for den målrettede forskning, som har fundet sted med såkaldte ’correctors’ og ’potentiators’ siden 1998. Læs mere om de nyeste behandlingsmuligheder her. LINK
Mutationer i klasse I, II og III betegnes generelt som de svære/alvorlige mutationer og dækker godt 90 % procent af alle patienter med cystisk fibrose.
Mutationerne i klasse I-III kan ikke transportere klorid-ioner (cl-) og bikarbonat over cellemembranen fra det indre til det ydre af cellevæggen, og patienter i disse klasser er generelt kendetegnet ved manglende eller nedsat produktion af fordøjelsesenzymer og lungekomplikationer.
Mutationer i klasse IV, V og VI betegnes generelt som de lette/mindre alvorlige klasser og dækker godt 10% af patienterne. Modsat klasse I-III mutationerne, kan mutationerne i klasse IV og V godt transportere klorid-ioner (cl-) og bikarbonat i større eller mindre omfang. Derfor har patienterne i disse to klasser væsentligt færre lunge- og fordøjelsesproblemer, sammenlignet med patienterne i klasse I-III.